Erbitux
Le Informazioni Presenti Sul Sito Non Costituiscono Consulenza Medica. Non Vendiamo Nulla. L'Accuratezza Della Traduzione Non È Garantita. Disclaimer
Riepilogo della droga
Cos'è Erbitux?
Erbitux (cetuximab) in combinazione con la radioterapia è un anticorpo monoclonale indicato per il trattamento iniziale del carcinoma della testa e del collo avanzato a livello locale o regionale di un tipo specifico (carcinoma a cellule squamose). Utilizzato da solo Erbitux è anche approvato per trattare i pazienti con tumori della testa e del collo che sono tornati nella stessa posizione o si sono diffusi in altre parti del corpo e per i tumori della testa e del collo che sono progrediti a seguito a base di platino chemioterapia . L'erbitux viene anche utilizzato su tumori del colon -retto metastatici che contengono recettori del fattore di crescita epidermico.
Quali sono gli effetti collaterali di Erbitux?
Gli effetti collaterali più comuni di Erbitux includono:
- eruzione cutanea
- prurito
- pelle secca o rotta
- Cambiamenti delle unghie
- mal di testa
- diarrea
- nausea
- vomito
- mal di stomaco
- perdita di peso
- debolezza e
- Infezioni respiratorie della pelle e della bocca.
L'erbitux può anche causare potassio e calcio a basso sangue. I pazienti che assumono erbitux dovrebbero limitare la loro esposizione al sole. Gli effetti collaterali rari ma gravi di Erbitux includono:
- reazioni allergiche potenzialmente letali e
- Attacchi di cuore soprattutto se il paziente stava ottenendo anche chemioterapia o radioterapia.
Cerca cure mediche o chiama il 911 contemporaneamente se hai i seguenti gravi effetti collaterali:
- Sintomi di occhiali gravi come la perdita di visione improvvisa del tunnel della visione sfocata Dolore alla visione o gonfiore o vedere aloni attorno alle luci;
- Sintomi cardiaci gravi come battiti cardiaci irregolari o martellanti veloci; svolazzando nel petto; fiato corto; e improvviso vertigini giuria o svenuta;
- Grave mal di testa confusione Il braccio del linguaggio bloccato o la debolezza delle gambe problemi perdite perdita di coordinamento sentendoti instabili muscoli molto rigidi ad alta febbre abbondante sudorazione o tremori.
Questo documento non contiene tutti i possibili effetti collaterali e altri possono verificarsi. Verificare con il tuo medico ulteriori informazioni sugli effetti collaterali.
Dosaggio per erbitux
L'erbitux viene fornito ad una concentrazione di 2 mg/mL in fiale monouso da 100 mg (50 mL) o 200 mg (100 mg). Il dosaggio e la somministrazione di Erbitux IV dovrebbero essere fatti solo da quelli addestrati nella somministrazione di questo farmaco.
Quali sostanze o integratori di farmaci interagiscono con Erbitux?
Erbitux può interagire con altri farmaci. Dì al medico tutti i farmaci e gli integratori che usi.
Erbitux durante la gravidanza e l'allattamento
Di 'al medico se sei incinta o hai intenzione di rimanere incinta durante l'utilizzo di Erbitux; Non è noto se danneggerà un feto. Uomini e donne dovrebbero Utilizzare il controllo delle nascite per prevenire la gravidanza durante la ricezione di Erbitux e per almeno 6 mesi dopo la fine del trattamento. Non è noto se Erbitux passi nel latte materno o se potrebbe danneggiare un bambino allattante. L'allattamento al seno non è raccomandato durante la ricezione di Erbitux e per almeno 60 giorni dopo la fine del trattamento.
Ulteriori informazioni
Il nostro centro farmacologico Erbitux Effects fornisce una visione completa delle informazioni disponibili sui farmaci sui potenziali effetti collaterali quando si assume questo farmaco.
Informazioni sui farmaci FDA
- Descrizione del farmaco
- Indicazioni
- Dosaggio
- Effetti collaterali
- Avvertimenti
- Overdose
- Farmacologia clinica
- Guida ai farmaci
AVVERTIMENTO
Reazioni di infusione gravi e arresto cardiopolmonare
Reazioni di infusione: gravi reazioni di infusione si sono verificate con la somministrazione di erbitux in circa il 3% dei pazienti in studi clinici con esiti fatali riportati in meno di 1 su 1000 [vedi avvisi e PRECAUZIONI Reazioni avverse ]. Interrompere immediatamente e interrompere permanentemente l'infusione di erbitux per gravi reazioni di infusione [vedi Dosaggio e amministrazione AVVERTIMENTOS AND PRECAUZIONI ]. Arresto cardiopolmonare: arresto cardiopolmonare e/o morte improvvisa si sono verificate nel 2% dei pazienti con carcinoma a cellule squamose della testa e del collo trattate con erbitux e radioterapia nello Studio 1 e nel 3% dei pazienti con carcinoma a livello squamoso del testa e del centesimo e del cognoma e del cognoma e del cognoma. Nello studio 2. Monitorare da vicino elettroliti sierici tra cui potassio sierico di potassio e calcio durante e dopo la somministrazione di Erbitux [vedi avvisi e avvertimenti e PRECAUZIONI Studi clinici ].
Descrizione per Erbitux
Erbitux® (cetuximab) è un anticorpo monoclonale chimerico ricombinante/topo che si lega specificamente al dominio extracellulare del recettore del fattore di crescita epidermico umano (EGFR). Cetuximab è composto dalle regioni FV di un anticorpo murino anti-EGFR con IgG1 umane IgG1 REGIONI COSTANTI CHILE LIGHT CHILE e ha un peso molecolare approssimativo di 152 kDa. Cetuximab è prodotto nella coltura cellulare dei mammiferi (mieloma murino). L'erbitux è un liquido incolore chiaro sterile di pH da 7,0 a 7,4 che può contenere una piccola quantità di particelle di cetuximab amorfo bianco facilmente visibili. L'erbitux viene fornito ad una concentrazione di 2 mg/mL in fiale monouso da 100 mg (50 mL) o 200 mg (100 mL). Il cetuximab è formulato in una soluzione senza conservanti che contiene 8,48 mg/ml di cloruro di sodio 1,88 mg/ml di sodio di sodio ettaidrato dibasico 0,41 mg/ml di sodio monoidrata monoidrato e acqua per l'iniezione.
Usi per Erbitux
Carcinoma a cellule squamose della testa e del collo (SCCHN)
Erbitux® è indicato:
- In combinazione con radioterapia per il trattamento iniziale del carcinoma a cellule squamose localmente o regionali della testa e del collo (SCCHN).
- In combinazione con terapia a base di platino con fluorouracile per il trattamento di prima linea di pazienti con malattia locoregionale ricorrente o SCCHN metastatico.
- Come singolo agente per il trattamento di pazienti con SCCHN ricorrente o metastatico per i quali la terapia a base di platino ha fallito.
Cancro del colon-retto che esprime EGFR di tipo selvaggio K-RAS (CRC)
L'erbitux è indicato per il trattamento del recettore del fattore di crescita epidermico di tipo selvaggio K-RAS (EGFR) che esprime il carcinoma del colon-retto metastatico (MCRC) determinato da un test approvato dalla FDA [vedi Dosaggio e amministrazione ]:
- in combinazione con Folfiri (Irinotecan Fluorouracil Leucovorin) per il trattamento di prima linea
- in combinazione con irinotecan in pazienti refrattari a base di irinotecan chemioterapia
- Come singolo agente nei pazienti che hanno fallito la chemioterapia a base di ossaliplatino e irinotecan o che sono intolleranti a Irinotecan.
Limiti di utilizzo
Erbitux non è indicato per il trattamento del carcinoma del colon-retto Mutante RAS o quando i risultati dei test di mutazione RAS sono sconosciuti [vedi AVVERTIMENTOS AND PRECAUZIONI ].
BRAF V600E Mutation-Positive Metastatic Cancer (CRC)
ERBITUX è indicato in combinazione con Encorafenib per il trattamento di pazienti adulti con carcinoma del colon-retto metastatico (CRC) con una mutazione BRAF V600E rilevata da un test approvato dalla FDA dopo una terapia precedente [vedi Dosaggio e amministrazione ].
Dosaggio per erbitux
Selezione del paziente
Seleziona i pazienti con carcinoma del colon -retto metastatico (CRC) per il trattamento con Erbitux in base alla presenza di:
- RAS wild wild-type EGFR che esprime CRC [vedi Studi clinici ] O
- BRAF V600E Mutation-positive Metastatic CRC [vedi Studi clinici ]
Le informazioni sui test approvati dalla FDA per il rilevamento di mutazioni K-RAS o BRAF V600E in CRC in pazienti con CRC metastatico sono disponibili su: https://www.fda.gov/companiondiagnostics.
Dosaggio raccomandato per carcinoma a cellule squamose della testa e del collo (SCCHN)
In combinazione con la radioterapia
- Dose iniziale: 400 mg/m² somministrato come infusione endovenosa di 120 minuti una settimana prima di iniziare un corso di radioterapia.
- Dosi successive: 250 mg/m² somministrato come infusione di 60 minuti ogni settimana per la durata della radioterapia (6 settimane di 7 settimane).
- Somministrazione Erbitux completa 1 ora prima della radioterapia.
Come un singolo agente o in combinazione con terapia a base di platino e fluorouracile
Somministrare Erbitux come singolo agente o in combinazione con terapia a base di platino e fluorouracile su un programma settimanale o bisettimanale.
Dosaggio settimanale
- Dose iniziale: 400 mg/m² somministrato come infusione endovenosa di 120 minuti
- Dosi successive: 250 mg/m² somministrato come infusione di 60 minuti ogni settimana
Dosaggio bisettimanale
- Dosi iniziali e successive: 500 mg/m² somministrato come infusione endovenosa di 120 minuti ogni 2 settimane
Somministrazione Erbitux completa 1 ora prima della terapia a base di platino con fluorouracile. Continuare il trattamento fino alla progressione della malattia o alla tossicità inaccettabile.
Dosaggio raccomandato per il cancro del colon -retto (CRC)
Come un singolo agente o in combinazione con irinotecan o folfiri (Irinotecan fluorouracil leucovorin)
Somministrare Erbitux come un singolo agente o in combinazione con Irinotecan o Folfiri (Irinotecan Fluorouracil Leucovorin) su un programma settimanale o bisettimanale.
Dosaggio settimanale
- Dose iniziale: 400 mg/m² somministrato come infusione endovenosa di 120 minuti
- Dosi successive: 250 mg/m² somministrato come infusione di 60 minuti ogni settimana
Dosaggio bisettimanale
- Dosi iniziali e successive: 500 mg/m² somministrato come infusione endovenosa di 120 minuti ogni 2 settimane
Completa Erbitux Administration 1 ora prima di Irinotecan o Folfiri. Continuare il trattamento fino alla progressione della malattia o alla tossicità inaccettabile
In combinazione con encorafenib
- La dose iniziale raccomandata è di 400 mg/m² somministrata come infusione endovenosa di 120 minuti in combinazione con Encorafenib.
- Il dosaggio successivo raccomandato è di 250 mg/m² a settimana come infusione di 60 minuti in combinazione con encorafenib fino alla progressione della malattia o alla tossicità inaccettabile.
Fare riferimento alle informazioni di prescrizione di Encorafenib per le informazioni di dosaggio di Encorafenib consigliate.
Premedicazione
Premedicate con un antagonista del recettore di istamina-1 (H1) per via endovenosa 30-60 minuti prima della prima dose o dosi successive come ritenuto necessario [vedi AVVERTIMENTOS AND PRECAUZIONI ].
Dosaggio Modifications For Reazione avversas
Ridurre il ritardo o interrompere l'erbitux per gestire le reazioni avverse come descritto nella Tabella 1.
Tabella 1: modifiche al dosaggio consigliate per le reazioni avverse
| Reazione avversa | Gravitàa | Dosaggio Modification |
| Reazioni di infusione [vedi AVVERTIMENTOS AND PRECAUZIONI ] | Grado 1 o 2 | Ridurre il tasso di infusione del 50%. |
| Grado 3 o 4 | Immediatamente e permanentemente interrompere l'erbitux. | |
| Tossicità dermatologiche e sequele infettive (ad es. Malattia mucocutanea cutanea acneiforme) [vedi AVVERTIMENTOS AND PRECAUZIONI ] | 1 ° occorrenza; Grado 3 o 4 | Ritardare l'infusione da 1 a 2 settimane; Se la condizione migliora, continua a 250 mg/m². Se nessun miglioramento interrompere l'erbitux. |
| 2a occorrenza; Grado 3 o 4 | Ritardare l'infusione da 1 a 2 settimane; Se la condizione migliora, continua a 200 mg/m². Se nessun miglioramento interrompere l'erbitux. | |
| 3 ° occorrenza; Grado 3 o 4 | Ritardare l'infusione da 1 a 2 settimane; Se la condizione migliora, continua a 150 mg/m². Se nessun miglioramento interrompere l'erbitux. | |
| 4 ° occorrenza; Grado 3 o 4 | Interrompere l'erbitux. | |
| Tossicità polmonare [vedi AVVERTIMENTOS AND PRECAUZIONI ] | Insorgenza acuta o peggioramento dei sintomi polmonari | Ritardare l'infusione da 1 a 2 settimane; Se le condizioni migliorano continuano alla dose che veniva somministrata al momento del verificarsi. Se nessun miglioramento in 2 settimane o la malattia polmonare interstiziale (ILD) viene confermato interrompere l'erbitux. |
| a National Cancer Institute (NCI) Criteri di tossicità comune (CTC) versione 2.0. |
Preparazione per l'amministrazione
- La soluzione dovrebbe essere chiara e incolore e può contenere una piccola quantità di particelle di cetuximab amorfo bianco facilmente visibili. Non agitare o diluire.
- Ispezionare visivamente il particolato estraneo e lo scolorimento prima dell'amministrazione ogni volta che la soluzione e il consumo del contenitore. Non utilizzare se la soluzione è scolorita nuvolosa o contiene particolato estraneo.
- Non somministrare Erbitux come spinta e bolo endovenosa.
- Somministrare tramite pompa di infusione o pompa a siringa. Non superare una velocità di infusione di 10 mg/min.
- Somministrare attraverso un filtro in linea 0,22 micrometro a basso legame.
Come fornito
Dosaggio Forms And Strengths
Iniezione: 100 mg/50 mL (2 mg/mL) o 200 mg/100 ml (2 mg/mL) come soluzione chiara incolore in una fiala monodose.
Iniezione Erbitux® (cetuximab) è una soluzione incolore chiara senza conservanti sterili in una fiala monodose da 2 mg/ml fornita come segue:
100 mg/50 ml confezionato individualmente in un cartone ( Ndc 66733-948-23)
200 mg/100 ml confezionato individualmente in un cartone ( Ndc 66733-958-23)
Archiviazione e maneggevolezza
- Conservare fiale in refrigerazione da 2 ° C a 8 ° C (da 36 ° F a 46 ° F).
- Non congelare o scuotere.
- Una maggiore formazione di particolato può verificarsi a temperature a 32 ° F) o inferiore a 0 ° C.
- Scartare qualsiasi soluzione rimanente nel contenitore di infusione dopo 8 ore a temperatura ambiente controllata o dopo 12 ore a 2 ° C a 8 ° C.
- Scartare qualsiasi parte inutilizzata della fiala.
Prodotta da IMCLONE LLC una consociata interamente di proprietà di Eli Lilly e della società Branchburg NJ 08876 USA Eli Lilly e la società Indianapolis nel 46285 USA. Revisionato: settembre 2021
Effetti collaterali per Erbitux
Le seguenti reazioni avverse sono discusse in maggior dettaglio in altre sezioni dell'etichetta:
- Reazioni di infusione [vedi AVVERTIMENTOS AND PRECAUZIONI ].
- Arresto cardiopolmonare [vedi AVVERTIMENTOS AND PRECAUZIONI ].
- Tossicità polmonare [vedi AVVERTIMENTOS AND PRECAUZIONI ].
- Tossicità dermatologica [vedi AVVERTIMENTOS AND PRECAUZIONI ].
- Anomalie di ipomagnesemia ed elettroliti [vedi AVVERTIMENTOS AND PRECAUZIONI ].
Esperienza di studi clinici
Poiché gli studi clinici sono condotti in condizioni di reazione avverse ampiamente variabili osservate negli studi clinici di un farmaco non possono essere paragonati direttamente ai tassi negli studi clinici di un altro farmaco e potrebbero non riflettere i tassi osservati nella pratica.
I dati descritti negli avvertimenti e nelle precauzioni riflettono l'esposizione a Erbitux in 1373 pazienti con SCCHN o CRC iscritti agli studi clinici e trattati al dosaggio raccomandato per una mediana da 7 a 14 settimane [vedi Studi clinici ]. The most common adverse reEtions in clinical trials with ERBITUX as a single-agent or in combination with radiotherapy or chemioterapia [Folfiri irinotecan and 5-fluorourEil/platinum] (incidence ≥25%) include cutaneous adverse reEtions (including eruzione cutanea pruritus and Cambiamenti delle unghie) mal di testa diarrea and infection.
Carcinoma a cellule squamose della testa e del collo (SCCHN)
In combinazione con la radioterapia
La sicurezza di Erbitux in combinazione con radioterapia rispetto alla sola radioterapia è stata valutata in Bonner. I dati descritti di seguito riflettono l'esposizione all'erbitux in 420 pazienti con SCCHN a livello locale o regionale. L'erbitux è stato somministrato al dosaggio raccomandato (400 mg/m² dose iniziale seguita da 250 mg/m² a settimana). I pazienti hanno ricevuto una mediana di 8 infusioni (intervallo da 1 a 11) [vedi Studi clinici ].
La tabella 2 fornisce la frequenza e la gravità delle reazioni avverse in Bonner.
Tabella 2: reazioni avverse selezionate in ≥10% dei pazienti con SCCHN locoregionalmente avanzato (Bonner)a
| Reazione avversa | Erbitux con radiazioni (n = 208) | Solo radioterapia (n = 212) | ||
| Gradi 1-4b | Gradi 3 e 4 | Gradi 1-4 | Gradi 3 e 4 | |
| Generale | ||||
| Astenia | 56 | 4 | 49 | 5 |
| Febbrec | 29 | 1 | 13 | 1 |
| Mal di testa | 19 | <1 | 8 | <1 |
| Brividic | 16 | 0 | 5 | 0 |
| Reazione di infusioned | 15 | 3 | 2 | 0 |
| Infezione | 13 | 1 | 9 | 1 |
| Gastrointestinale | ||||
| Nausea | 49 | 2 | 37 | 2 |
| Emesi | 29 | 2 | 23 | 4 |
| Diarrea | 19 | 2 | 13 | 1 |
| Dispepsia | 14 | 0 | 9 | 1 |
| Metabolismo e nutrizione | ||||
| Perdita di peso | 84 | 11 | 72 | 7 |
| Disidratazione | 25 | 6 | 19 | 8 |
| Aumento della transaminasi alaninae | 43 | 2 | 21 | 1 |
| Aumento della transaminasi aspartatae | 38 | 1 | 24 | 1 |
| Aumentata fosfatasi alcalina | 33 | <1 | 24 | 0 |
| Respiratorio | ||||
| Faringite | 26 | 3 | 19 | 4 |
| Dermatologico | ||||
| Eruzione cutanea acneiformef | 87 | 17 | 10 | 1 |
| Dermatite da radiazioni | 86 | 23 | 90 | 18 |
| Reazione del sito dell'applicazione | 18 | 0 | 12 | 1 |
| Prurito | 16 | 0 | 4 | 0 |
| a Reazioni avverse che si verificano in ≥10% dei pazienti nel braccio di combinazione Erbitux e ad un'incidenza più elevata (≥5%) rispetto al braccio da sola radiazione. b Le reazioni avverse sono state classificate utilizzando la versione 2.0 NCI CTC. c Include i casi riportati anche come reazione di infusione. d Reazione di infusione definita come qualsiasi evento descritto in qualsiasi momento durante lo studio clinico come reazione allergica o reazione anafilattoide o qualsiasi evento che si verifica il primo giorno di dosaggio descritto come reazione allergica di reazione anafilattoide Febbre brividi e febbre o dispnea. e Basato su misurazioni di laboratorio non su reazioni avverse riportate, il numero di soggetti con campioni testati variava da 205-206 per ERBITUX con braccio di radiazione; 209-210 solo per radiazioni. f Eruzione cutanea acneiforme definita come qualsiasi evento descritto come cutanea maculopapolare di acne cutanea pustolosa cutanea secca o dermatite esfoliativa. |
L'incidenza complessiva delle tossicità delle radiazioni tardive (qualsiasi grado) era più elevata per i pazienti che ricevevano Erbitux in combinazione con radioterapia rispetto alla sola radioterapia. Sono stati interessati i seguenti siti: ghiandole salivari (65% contro 56%) laringe (52% contro 36%) tessuto sottocutaneo (49% contro 45%) membrana mucosa (48% contro 39%) esofago (44% contro 35%) (42% contro 33%). L'incidenza delle tossicità delle radiazioni tardive di grado 3 o 4 era simile
tra la sola radioterapia e l'erbitux con gruppi di trattamento delle radiazioni.
In combinazione con terapia a base di platino e fluorouracile
La sicurezza di un prodotto cetuximab in combinazione con terapia a base di platino e terapia a base di fluorouracile o platino e solo fluorouracile è stata valutata in estrema. I dati descritti di seguito riflettono l'esposizione a un prodotto cetuximab in 434 pazienti con malattia locoregionale ricorrente o SCCHN metastatico. Poiché ERBITUX fornisce un'esposizione più alta del 22% rispetto al prodotto cetuximab, i dati forniti di seguito possono sottovalutare l'incidenza e la gravità delle reazioni avverse previste con ERbitux per questa indicazione; Tuttavia, la tollerabilità della dose raccomandata è supportata da dati di sicurezza da ulteriori studi su ERbitux [vedi Farmacologia clinica ]. Cetuximab was administered intravenously at a dosage of 400 mg/m² for the initial dose followed by 250 mg/m² weekly. Patients received a median of 17 infusions (range 1 to 89) [see Studi clinici ].
La tabella 3 fornisce la frequenza e la gravità delle reazioni avverse in estrema.
Tabella 3: reazioni avverse selezionate in ≥10% dei pazienti con malattia locoregionale ricorrente o SCCHN metastatico (Extreme)a
| Reazione avversa | Cetuximab con terapia a base di platino e fluorouracile (n = 219) | Terapia a base di platino e solo fluorocile (n = 215) | ||
| Gradi 1-4b | Gradi 3 e 4 | Gradi 1-4 | Gradi 3 e 4 | |
| Occhio | ||||
| Congiuntivite | 10 | 0 | 0 | 0 |
| Gastrointestinale | ||||
| Nausea | 54 | 4 | 47 | 4 |
| Diarrea | 26 | 5 | 16 | 1 |
| Generale and Administration Site | ||||
| Pyrexia | 22 | 0 | 13 | 1 |
| Reazione di infusionec | 10 | 2 | <1 | 0 |
| Infeziones | ||||
| Infezioned | 44 | 11 | 27 | 8 |
| Metabolismo e nutrizione | ||||
| Anoressia | 25 | 5 | 14 | 1 |
| Ipocalcemia | 12 | 4 | 5 | 1 |
| Iponokalemia | 12 | 7 | 7 | 5 |
| Ipomagnesemia | 11 | 5 | 5 | 1 |
| Dermatologico | ||||
| Eruzione cutanea acneiformee | 70 | 9 | 2 | 0 |
| Eruzione cutanea | 28 | 5 | 2 | 0 |
| Acne | 22 | 2 | 0 | 0 |
| Dermatite acneiforme | 15 | 2 | 0 | 0 |
| Pelle secca | 14 | 0 | <1 | 0 |
| Alopecia | 12 | 0 | 7 | 0 |
| a Reazioni avverse che si verificano in ≥10% dei pazienti nel braccio di combinazione di cetuximab e ad un'incidenza più elevata (≥5%) rispetto alla terapia a base di platino e al solo braccio di fluorocile. b Le reazioni avverse sono state classificate utilizzando la versione 2.0 NCI CTC. c Reazione di infusione definita come febbre di ipersensibilità di reazione anafilattica e/o brividi dispnea o piressia il primo giorno di dosaggio. d Infezione excludes sepsis-related events which are presented separately. e Acneiform eruzione cutanea defined as Ene dermatitis Eneiform dry skin exfoliative eruzione cutanea eruzione cutanea eruzione cutanea erythematous eruzione cutanea mEular eruzione cutanea papular or eruzione cutanea pustular. Chemioterapia = cisplatino e fluorouracile o carboplatino e fluorouracile |
Per i disturbi cardiaci circa il 9% dei pazienti in entrambi i bracci di trattamento in Extreme ha avuto un evento cardiaco. La maggior parte di questi eventi si è verificata in pazienti che hanno ricevuto cisplatino e fluorouracile con o senza cetuximab. Sono stati osservati disturbi cardiaci nell'11% e nel 12% dei pazienti che hanno ricevuto rispettivamente cisplatino e fluorouracile con o senza cetuximab e 6% e 4% in pazienti che hanno ricevuto carboplatina e fluorouracile rispettivamente con o senza cetuximab. In entrambe le armi l'incidenza di eventi cardiovascolari era più elevata nel cisplatino e nel fluorouracile contenente sottogruppo. La morte attribuita agli eventi cardiovascolari o alla morte improvvisa è stata riportata nel 3% dei pazienti nel cetuximab con terapia a base di platino e braccio di fluorouracile e nel 2% dei pazienti nella terapia a base di platino e nel solo braccio di fluorocile.
K-RAS wild-type che esprime il carcinoma del colon-retto metastatico (MCRC)
In combinazione con folfiri
La sicurezza di un prodotto cetuximab in combinazione con Folfiri o Folfiri da solo è stata valutata in cristallo. I dati descritti di seguito riflettono l'esposizione a un prodotto Cetuximab in 667 pazienti con MCRC che esprimono EGFR K-RAS. Erbitux fornisce un'esposizione più alta del 22% rispetto a questo prodotto; Tuttavia, i dati di sicurezza di Crystal sono coerenti nell'incidenza e nella gravità delle reazioni avverse con quelle osservate per Erbitux in questa indicazione. Cetuximab è stato somministrato per via endovenosa ad un dosaggio di 400 mg/m² di dose iniziale seguita da 250 mg/m² a settimana. I pazienti hanno ricevuto una mediana di 24 infusioni (intervallo da 1 a 224) [vedi Studi clinici ].
Le gravi reazioni avverse includevano l'embolismo polmonare che è stato riportato nel 4,4% dei pazienti trattati con cetuximab con FOLFIRI rispetto al 3,4% dei pazienti trattati con solo con FOLFIRI.
La tabella 4 fornisce la frequenza e la gravità delle reazioni avverse nel cristallo.
Tabella 4: reazioni avverse selezionate in ≥10% dei pazienti con carcinoma del colon-retto wild-type ed EGFR che esprimono EGFR (cristallo)a
| Reazione avversa | Cetuximab con folfiri (n = 317) | Folforing da solo (n = 350) | ||
| Gradi 1-4b | Gradi 3 e 4 | Gradi 1-4 | Gradi 3 e 4 | |
| Ematologico | ||||
| Neutropenia | 49 | 31 | 42 | 24 |
| Occhio | ||||
| Congiuntivite | 18 | <1 | 3 | 0 |
| Gastrointestinale | ||||
| Diarrea | 66 | 16 | 60 | 10 |
| Stomatite | 31 | 3 | 19 | 1 |
| Dispepsia | 16 | 0 | 9 | 0 |
| Generale and Administration Site | ||||
| Pyrexia | 26 | 1 | 14 | 1 |
| Il peso è diminuito | 15 | 1 | 9 | 1 |
| Reazione di infusionec | 14 | 2 | <1 | 0 |
| Infeziones | ||||
| Parornia | 20 | 4 | <1 | 0 |
| Metabolismo e nutrizione | ||||
| Anoressia | 30 | 3 | 23 | 2 |
| Dermatologico | ||||
| Acne-like Eruzione cutanead | 86 | 18 | 13 | <1 |
| Eruzione cutanea | 44 | 9 | 4 | 0 |
| Dermatite acneiforme | 26 | 5 | <1 | 0 |
| Pelle secca | 22 | 0 | 4 | 0 |
| Acne | 14 | 2 | 0 | 0 |
| Prurito | 14 | 0 | 3 | 0 |
| Sindrome di Eritrodisestesia Palmar-Plantar | 19 | 4 | 4 | <1 |
| Fessure cutanee | 19 | 2 | 1 | 0 |
| a Reazioni avverse che si verificano in ≥10% dei pazienti nel braccio di combinazione di cetuximab e ad un'incidenza più elevata (≥5%) rispetto al braccio solo Folfiri. b Le reazioni avverse sono state classificate utilizzando la versione 2.0 NCI CTC. c Reazione di infusione definita come qualsiasi evento che incontra i concetti medici di allergia/anafilassi in qualsiasi momento durante lo studio clinico o qualsiasi evento che si verifica il primo giorno di dosaggio e che incontra i concetti medici di dispnea e febbre della pressione emocolta del sangue. Insufficienza cardiovascolare Clonus Convulsion Convulsione coronarica non fenomeno Epilessia Ipertensione Ipertensione Ipertensiva Crisi ipertensiva di emergenza Ipotensione La perdita di reazione di reazione di coscienza dell'ischemia miocardica miocardica dell'anga-shock di anne-shock improvviso della morte o ipertensione sistolica. d Acne-like eruzione cutanea defined by the following events: Ene Ene pustular butterfly eruzione cutanea dermatitis Eneiform drug eruzione cutanea with eosinophilia and systemic symptoms dry skin erythema exfoliative eruzione cutanea folliculitis genital eruzione cutanea mucocutaneous eruzione cutanea pruritus eruzione cutanea eruzione cutanea erythematous eruzione cutanea follicular eruzione cutanea generalized eruzione cutanea mEular eruzione cutanea mEulopapular eruzione cutanea mEulovesicular eruzione cutanea morbilliform eruzione cutanea papular eruzione cutanea papulosquamous eruzione cutanea pruritic eruzione cutanea pustular eruzione cutanea rubelliform eruzione cutanea scarlatiniform eruzione cutanea vesicular skin exfoliation skin hyperpigmentation skin plaque telangiectasia or xerosis. |
Come singolo agente
La sicurezza di Erbitux con la migliore cure di supporto (BSC) o BSC da sola è stata valutata nello studio CA225-025. I dati descritti di seguito riflettono l'esposizione all'erbitux in 242 pazienti con carcinoma del colon-retto metastatico che esprimono EGFR K-RAS (MCRC) [vedi AVVERTIMENTOS AND PRECAUZIONI ]. ERBITUX was administered intravenously at the recommended dosage (400 mg/m² initial dose followed by 250 mg/m² weekly). Patients received a median of 17 infusions (range 1 to 51) [see Studi clinici ].
La tabella 5 fornisce la frequenza e la gravità delle reazioni avverse nello studio CA225-025.
Tabella 5: reazioni avverse selezionate in ≥10% dei pazienti con carcinoma del colon-retto metastatico che esprimono EGFR K-RAS trattato con erbitux singolo (studio CA225-025)a
| Reazione avversa | Erbitux con BSC (n = 118) | BSC da solo (n = 124) | ||
| Gradi 1-4b | Gradi 3 e 4 | Gradi 1-4 | Gradi 3 e 4 | |
| Dermatologico | ||||
| Eruzione cutanea/Desquamation | 95 | 16 | 21 | 1 |
| Pelle secca | 57 | 0 | 15 | 0 |
| Prurito | 47 | 2 | 11 | 0 |
| Altra dermatologia | 35 | 0 | 7 | 2 |
| Cambiamenti delle unghie | 31 | 0 | 4 | 0 |
| Generale | ||||
| Fatica | 91 | 31 | 79 | 29 |
| Febbre | 25 | 3 | 16 | 0 |
| Reazione di infusionesc | 18 | 3 | 0 | 0 |
| Rigilli rigidi | 16 | 1 | 3 | 0 |
| Dolore | ||||
| Dolore-Altro | 59 | 18 | 37 | 10 |
| Mal di testa | 38 | 2 | 11 | 0 |
| Dolore alle ossa | 15 | 4 | 8 | 2 |
| Polmonare | ||||
| Dispnea | 49 | 16 | 44 | 13 |
| Tosse | 30 | 2 | 19 | 2 |
| Gastrointestinale | ||||
| Nausea | 64 | 6 | 50 | 6 |
| Stipsi | 53 | 3 | 38 | 3 |
| Diarrea | 42 | 2 | 23 | 2 |
| Vomito | 40 | 5 | 26 | 5 |
| Stomatite | 32 | 1 | 10 | 0 |
| Altro | 22 | 12 | 16 | 5 |
| Disidratazione | 13 | 5 | 3 | 0 |
| Secchezza della bocca | 12 | 0 | 6 | 0 |
| Disturbo del gusto | 10 | 0 | 5 | 0 |
| Infezione | ||||
| Infezione without neutropenia | 38 | 11 | 19 | 5 |
| Muscoloscheletrico | ||||
| Artralgia | 14 | 3 | 6 | 0 |
| Neurologico | ||||
| Neuropatia-sensoriale | 45 | 1 | 38 | 2 |
| Insonnia | 27 | 0 | 13 | 0 |
| Confusione | 18 | 6 | 10 | 2 |
| Ansia | 14 | 1 | 5 | 1 |
| Depressione | 14 | 0 | 5 | 0 |
| a Reazioni avverse che si verificano in ≥10% dei pazienti nell'erbitux con braccio BSC e ad un'incidenza più elevata (≥5%) rispetto al braccio da solo BSC. b Le reazioni avverse sono state classificate utilizzando la versione 2.0 NCI CTC. c Reazione di infusione definita come qualsiasi evento (i brividi rigorono la dispnea tachicardia broncospasmo per la tensione del torace gonfiore dell'ipotensione di ipotensione di eruzione cutanea nausea angoedema dolore che suda tremori che scuotono la febbre farmacologica o altri reazioni di ipersensibilità) registrato dall'investigatore come correlato all'infusione. |
In combinazione con irinotecan
L'erbitux al dosaggio raccomandato è stato somministrato in combinazione con irinotecan in 354 pazienti con MCRC ricorrente EGFrexpressing nello studio CP02-9923 e il legame.
Le reazioni avverse più comuni erano l'eruzione acneiforme (88%) astenia/malessere (73%) diarrea (72%) e nausea (55%). I gradi più comuni 3-4 reazioni avverse includevano la diarrea (22%) leucopenia (17%) Ashenia/Malaise (16%) ed eruzione acneiforme (14%).
BRAF V600E Mutation-Positive Metastatic Cancer (CRC) In combinazione con encorafenib
La sicurezza di Erbitux (400 mg/m² dose iniziale seguita da 250 mg/m² a settimana) in combinazione con Encorafenib (300 mg una volta al giorno) è stata valutata in 216 pazienti con CRC metastatico positivo alla mutazione BRAF V600E in uno studio randomizzato con apertura attivo (BEACON CRC). Il processo Beacon CRC [vedi Studi clinici ] hanno escluso i pazienti con una storia della sindrome di Gilbert anormale frazione di eiezione ventricolare sinistra prolungata QTC (> 480 ms) ipertensione non controllata e storia o prove attuali dell'occlusione della vena retinica. La durata mediana dell'esposizione è stata di 4,4 mesi per i pazienti trattati con Erbitux in combinazione con Encorafenib e 1,6 mesi per i pazienti trattati con Irinotecan o 5-Fluorouracile (5-FU)/Acido folinico (FA)/IRINOTECAN (FOLFIRI) in combinazione con ERBITUX.
Le reazioni avverse più comuni (≥25%) nei pazienti che ricevevano erbitux in combinazione con encorafenib erano la dermatite da diarrea dermatite da dermatite acneiforme di nausea fatica che ridotte l'artralgia e l'eruzione.
La Tabella 6 e la Tabella 7 presentano reazioni avverse ai farmaci e anomalie di laboratorio identificate rispettivamente in Beacon CRC.
Tabella 6: reazioni avverse che si verificano in ≥10% dei pazienti che hanno ricevuto erbitux in combinazione con Encorafenib in Beacon CRCa
| Reazione avversa | Erbitux con encorafenib N = 216 | Erbitux con irinotecan o erbitux con folfiri N = 193 | ||
| Tutti i gradi (%) | ≥ Grado 3b (%) | Tutti i gradi (%) | ≥ Grado 3 (%) | |
| Generale Disorders and Administration Site Conditions | ||||
| Faticac | 51 | 7 | 50 | 8 |
| Pyrexiac | 17 | 1 | 15 | 1 |
| Gastrointestinale Disorders | ||||
| Nausea | 34 | 1 | 41 | 1 |
| Diarreac | 33 | 2 | 48 | 10 |
| Dolore addominalec | 30 | 4 | 32 | 5 |
| Vomito | 21 | 1 | 29 | 3 |
| Stipsi | 15 | 0 | 18 | 1 |
| Metabolismo e nutrizione Disorders | ||||
| Diminuzione dell'appetito | 27 | 2 | 27 | 3 |
| Muscoloscheletrico and Connective Tissue Disorders | ||||
| Artralgiac | 27 | 4 | 3 | 0 |
| Miopatiac | 15 | 1 | 4 | 0 |
| Dolore in extremity | 10 | 0 | 1 | 0 |
| Disturbi della pelle e dei tessuti sottocutanei | ||||
| Dermatite acneiformec | 32 | 1 | 43 | 3 |
| Eruzione cutaneac | 26 | 0 | 26 | 2 |
| Pruritoc | 14 | 0 | 6 | 0 |
| Nevus melanocitico | 14 | 0 | 0 | 0 |
| Pelle seccac | 13 | 0 | 12 | 1 |
| Disturbi del sistema nervoso | ||||
| Mal di testac | 20 | 0 | 3 | 0 |
| Neuropatia perifericac | 12 | 1 | 6 | 0 |
| Disturbi vascolari | ||||
| Emorragiac | 19 | 2 | 9 | 0 |
| Disturbi psichiatrici | ||||
| Insonniac | 13 | 0 | 6 | 0 |
| a Gradi per National Cancer Institute CTCAE V4.03. b Le reazioni avverse di grado 4-5 nell'erbitux con braccio Encorafenib erano limitate all'emorragia di grado 5 (n = 1). c Rappresenta un composito di più termini preferiti correlati. |
Altro clinically important adverse reEtions occurring in <10% of patients who received ERBITUX in combination with encorDienib were:
Gastrointestinale disorders: Pancreatitis
Tabella 7: anomalie di laboratorio che si verificano in ≥10% (tutti i gradi) di pazienti che hanno ricevuto erbitux in combinazione con Encorafenib in Beacon CRCa
| Anomalia di laboratoriob | Erbitux con encorafenib | Erbitux con irinotecan con o erbitux con folfiri | ||
| Tutti i gradi (%) | Gradi 3 e 4 (%) | Tutti i gradi (%) | Gradi 3 e 4 (%) | |
| Ematologia | ||||
| Anemia | 34 | 4 | 48 | 5 |
| Linfopenia | 24 | 7 | 35 | 5 |
| Aumento del tempo di tromboplastina parziale attivato | 13 | 1 | 7 | 1 |
| Chimica | ||||
| Ipomagnesemia | 19 | 0 | 22 | 1 |
| Aumento della fosfatasi alcalina | 18 | 4 | 30 | 7 |
| Aumento alt | 17 | 0 | 29 | 3 |
| Aumento AST | 15 | 1 | 22 | 2 |
| Iponokalemia | 12 | 3 | 32 | 5 |
| Iponatriemia | 11 | 2 | 13 | 2 |
| a Gradi per National Cancer Institute CTCAE V4.03. b In base al numero di pazienti con basale disponibile e almeno un test di laboratorio sul trattamento. |
Immunogenicità
Come per tutte le proteine terapeutiche, esiste un potenziale per l'immunogenicità. Il rilevamento della formazione di anticorpi dipende fortemente dalla sensibilità e dalla specificità del dosaggio. Inoltre, l'incidenza osservata dell'anticorpo (compresa l'anticorpo neutralizzante) in un test può essere influenzata da diversi fattori tra cui la metodologia del test, i tempi di gestione dei campioni dei farmaci concomitanti di raccolta del campione e la malattia sottostante. Per questi motivi il confronto dell'incidenza di anticorpi con cetuximab negli studi seguenti con l'incidenza di anticorpi a cetuximab in altri studi o con altri prodotti può essere fuorviante.
È stata utilizzata una metodologia ELISA per caratterizzare l'incidenza di anticorpi anti-Cetuximab. L'incidenza di anticorpi leganti anticoluximab in 105 pazienti (dagli studi I4E-MC-JXBA I4E-MC-JXBB e I4E-MC-JXBD) con almeno un campione di sangue post-baseline (≥4 settimane dopo la prima somministrazione di Erbitux) era <5%.
Esperienza post -marketing
Le seguenti reazioni avverse sono state identificate durante l'uso post di approvazione di Erbitux. Poiché queste reazioni sono riportate volontariamente da una popolazione di dimensioni incerte, non è sempre possibile stimare in modo affidabile la loro frequenza o stabilire una relazione causale con l'esposizione ai farmaci.
- Neurologico: meningite asettica
- Gastrointestinale: Mucosal inflammation
- Dermatologico: Stevens-Johnson syndrome toxic epidermal necrolysis life-threatening and fatal bullous mucocutaneous disease
Interazioni farmacologiche per Erbitux
Nessuna informazione fornita
Avvertimenti per Erbitux
Incluso come parte del PRECAUZIONI sezione.
Precauzioni per Erbitux
Reazione di infusiones
L'erbitux può causare reazioni di infusione gravi e fatali. Le reazioni di infusione di qualsiasi grado si sono verificate nell'8,4% di 1373 pazienti che hanno ricevuto erbitux attraverso studi clinici. Reazioni di infusione gravi (gradi 3 e 4) si sono verificate nel 2,2% dei pazienti [vedi Reazioni avverse ]. Signs and symptoms included rapid onset of airway obstruction (bronchospasm stridor hoarseness) hypotension shock loss of consciousness myocardial infarction and/or cardiE arrest.
Il rischio di reazioni anafilattiche può essere aumentato nei pazienti con una storia di morsi di zecche allergia alla carne rossa o in presenza di anticorpi IgE diretti contro il galattosio-α-13-galattosio (alfa-gal). Considera di testare i pazienti per gli anticorpi IgE alfa-gal usando metodi a riposo FDA prima di iniziare l'erbitux. I risultati negativi per gli anticorpi alfa-gal non escludono il rischio di reazioni di infusione gravi.
Circa il 90% delle gravi reazioni di infusione si è verificato con la prima infusione nonostante la premedicazione con antistaminici. Le reazioni di infusione possono verificarsi durante o diverse ore dopo il completamento dell'infusione. Premedicate con un antagonista del recettore dell'istamina-1 (H1) come raccomandato [vedi Dosaggio e amministrazione ]. Monitor patients for at least 1 hour following eEh ERBITUX infusion in a setting with resuscitation equipment and other agents necessary to treat anaphylaxis. In patients requiring treatment for infusion reEtions monitor for more than 1 hour to confirm resolution of the reEtion. Interrupt the infusion and upon recovery resume the infusion at a slower rate or permanently discontinue ERBITUX based on severity [see Dosaggio e amministrazione ].
Arresto cardiopolmonare
Erbitux può causare un arresto cardiopolmonare. L'arresto cardiopolmonare o la morte improvvisa si sono verificati nel 2% di 208 pazienti trattati con radioterapia ed Erbitux a Bonner. Tre pazienti con precedente storia di malattia coronarica sono morti a casa con infarto del miocardio come presunta causa di morte. Uno di questi pazienti aveva aritmia e uno aveva insufficienza cardiaca congestizia. La morte si è verificata 27 32 e 43 giorni dopo l'ultima dose di Erbitux. Un paziente senza una precedente storia di malattia coronarica è morto un giorno dopo l'ultima dose di Erbitux.
In disturbi cardiaci fatali estremi e/o morte improvvisa si sono verificate nel 3% di 219 pazienti trattati con un prodotto Cetuximab in combinazione con terapia a base di platino e fluorocile.
Considerare attentamente l'uso di ERITUX con radioterapia o terapia a base di platino con fluorouracile in pazienti con SCCHN con una storia di insufficienza cardiaca congestizia o aritmie congestizia di arteria coronarica. Monitorare elettroliti sierici inclusi potassio sierico di potassio e calcio durante e dopo erbitux [vedi AVVERTIMENTOS AND PRECAUZIONI ].
Polmonare Toxicity
L'erbitux può causare malattie polmonari interstiziali (ILD). ILD tra cui 1 fatalità si è verificata in <0.5% of 1570 patients receiving ERBITUX in clinical trials.
Monitorare i pazienti per segni e sintomi di tossicità polmonare. Interrompere o interrompere permanentemente Erbitux per insorgenza acuta o peggioramento dei sintomi polmonari. Interrompere in modo permanente l'erbitux per l'ILD confermato [vedi Dosaggio e amministrazione ].
Dermatologico Toxicity
L'erbitux può causare tossicità dermatologiche tra cui essiccazione cutanea erutticata acneiforme e infiammazione paroniale infiattosa sequele infettive (ad esempio S. aureus Formazione di ascesso di sepsi Cellulite blefarite congiuntivite chetetite/cheratite ulcerosa con riduzione dell'acuità visiva Cheilite) e ipertricosi.
Acneiform eruzione cutanea occurred in 82% of the 1373 patients who received ERBITUX Eross clinical trials. Severe (Grades 3 or 4) Eneiform eruzione cutanea occurred in 10% of patients [see Reazioni avverse ]. Acneiform eruzione cutanea usually developed within the first two weeks of therapy; the eruzione cutanea lasted more than 28 days Diter stopping ERBITUX in most patients.
Malattia mucocutanea bollosa potenzialmente letale e fatale con erosioni di vesciche e sloughing cutaneo è stato osservato nei pazienti che hanno ricevuto erbitux. Non si può determinare se queste reazioni avverse mucocutanee fossero direttamente correlate all'inibizione dell'EGFR o agli effetti immuno-correlati idiosincratici (ad esempio sindrome di Stevens-Johnson o necrolisi epidermica tossica).
Monitorare i pazienti che ricevono erbitux per tossicità dermatologiche e sequele infettive. Istruire i pazienti a limitare l'esposizione al sole durante la terapia Erbitux. Trattenere la dose o interrompere permanentemente l'erbitux in base alla gravità dell'eruzione acneiforme o della malattia mucocutanea [vedi Dosaggio e amministrazione ].
Rischi associati all'uso in combinazione con radiazioni e cisplatino
In uno studio controllato 940 pazienti con SCCHN localmente avanzato sono stati randomizzati 1: 1 per ricevere l'erbitux in combinazione con radioterapia e cisplatino o radioterapia e solo cisplatino. L'aggiunta di ERBITUX ha comportato un aumento dell'incidenza della sindrome da richiamo radiazione di radiazione di grado 3 e 4 eventi cardiaci acneiformi e disturbi dell'elettroliti rispetto alle radiazioni e al solo cisplatino. Le reazioni avverse con esito fatale sono state riportate nel 4% dei pazienti nel braccio di combinazione Erbitux e nel 3% nel braccio di controllo. Nel braccio Erbitux il 2% ha sperimentato ischemia miocardica rispetto allo 0,9% nel braccio di controllo. Il principale risultato di efficacia dello studio è stato la sopravvivenza libera da progressione (PFS). L'aggiunta di Erbitux alle radiazioni e al cisplatino non ha migliorato la PFS. L'erbitux non è indicato per il trattamento di SCCHN in combinazione con radiazioni e cisplatino.
Ipomagnesemia And Accompanying Electrolyte Abnormalities
Erbitux può causare ipomagnesemia. L'ipomagnesemia si è verificata nel 55% di 365 pazienti che ha ricevuto ERbitux nello studio CA225-025 e altri due studi clinici in pazienti con carcinoma del colon-retto (CRC) o carcinoma della testa e del collo, compresi i gradi 3 e 4 nel 6% al 17%.
In estrema in cui un prodotto Cetuximab è stato somministrato in combinazione con terapia a base di platino l'aggiunta di cetuximab al cisplatino e al fluorocile ha comportato una maggiore incidenza di ipomagnesemia di qualsiasi grado (14%) e di ipomagnesemia di grado 3 o 4 (7%). L'ipomagnesemia di qualsiasi grado si è verificata nel 4% dei pazienti che hanno ricevuto carboplatina e fluorouracile di cetuximab. Nessun paziente ha sperimentato ipomagnesemia di grado 3 o 4 [vedi Reazioni avverse ].
Ipomagnesemia and Ecompanying electrolyte abnormalities can occur days to months Diter initiating ERBITUX. Monitor patients weekly during treatment for hypomagnesemia hypocalcemia and hypokalemia and for at least 8 weeks following the completion of ERBITUX. Replete electrolytes as necessary.
Aumento della progressione del tumore Aumento della mortalità o della mancanza di benefici nei pazienti con MCRC mutante RAS
Erbitux non è indicato per il trattamento di pazienti con CRC che ospitano mutazioni somatiche nell'esone 2 (codoni 12 e 13) esone 3 (codoni 59 e 61) e esone 4 (codoni 117 e 146) di K-RAS o N-RAS e di seguito è indicato come RAS o quando lo stato RAS è sconosciuto.
Sono state condotte analisi di sottoinsieme retrospettive di popolazioni di tipi di tipo RAS e di tipo selvaggio in diversi studi clinici randomizzati tra cui il cristallo per studiare il ruolo delle mutazioni di RAS sugli effetti clinici degli anticorpi monoclonali diretti anti-EGFR. L'uso di cetuximab nei pazienti con mutazioni RAS non ha comportato alcun beneficio clinico con la tossicità correlata al trattamento. Conferma lo stato della mutazione RAS nei campioni tumorali prima di iniziare ERbitux [vedi Indicazioni e utilizzo ].
Tossicità dell'embrione-fetale
Sulla base dei dati sugli animali e del suo meccanismo di azione ERBETUX possono causare danni fetali quando somministrato a una donna incinta. Non ci sono dati disponibili per l'esposizione a Erbitux nelle donne in gravidanza. In uno studio sulla riproduzione degli animali, la somministrazione endovenosa di cetuximab una volta a settimana a scimmie di cinomolgus in gravidanza durante il periodo di organogenesi ha provocato una maggiore incidenza di embrioletalità e aborto. L'interruzione o l'esaurimento dell'EGFR nei modelli animali provoca una compromissione dello sviluppo di embrioni, inclusi gli effetti sulla pelle cardiaca polmonare placentare e sullo sviluppo neurale. Consiglia alle donne in gravidanza del potenziale rischio per un feto. Consiglia alle femmine del potenziale riproduttivo di utilizzare una contraccezione efficace durante il trattamento con Erbitux e per 2 mesi dopo l'ultima dose di Erbitux [vedi Utilizzare in popolazioni specifiche ].
Tossicologia non clinica
Mutagenesi della carcinogenesi compromissione della fertilità
Non sono stati condotti studi sugli animali a lungo termine per testare il cetuximab per il potenziale cancerogeno e nessun potenziale mutageno o clastogenico del cetuximab è stato osservato nel test della Salmonella-Escherichia coli (AMES) o nel test del micronucleo del ratto in vivo. La cicalicità mestruale è stata compromessa nelle scimmie cinomolgus femminili che hanno ricevuto dosi settimanali da 0,4 a 4 volte la dose raccomandata di Erbitux (basata sulla superficie corporea totale). Gli animali trattati con cetuximab hanno mostrato un aumento delle incidenze di cicli irregolari o assenti rispetto agli animali di controllo. Questi effetti sono stati inizialmente annotati a partire dalla settimana 25 e sono continuati durante il periodo di recupero di 6 settimane. Non sono stati osservati effetti di cetuximab sui parametri misurati della fertilità maschile (ovvero i livelli sierici di testosterone e l'analisi dei conteggi degli spermatozoi di vitalità e motilità) rispetto al controllo delle scimmie maschili.
Utilizzare in popolazioni specifiche
Gravidanza
Riepilogo del rischio
Basato sui risultati degli studi sugli animali e sul suo meccanismo d'azione [vedi Farmacologia clinica ] L'erbitux può causare danni fetali quando somministrato a una donna incinta. Non ci sono dati disponibili per l'esposizione a Erbitux nelle donne in gravidanza. In uno studio sulla riproduzione degli animali, la somministrazione endovenosa di cetuximab una volta a settimana a scimmie di cinomolgus in gravidanza durante il periodo di organogenesi ha provocato una maggiore incidenza di embrioletalità e aborto. L'interruzione o l'esaurimento dell'EGFR nei modelli animali provoca una compromissione dello sviluppo di embrioni, inclusi gli effetti sulla pelle cardiaca polmonare placentare e sullo sviluppo neurale (vedi Dati ). È noto che le IgG umane attraversano la barriera placentare; Pertanto il cetuximab può essere trasmesso dalla madre al feto in via di sviluppo. Consiglia alle donne in gravidanza del potenziale rischio per un feto.
Nella popolazione generale degli Stati Uniti il rischio di background stimato di gravi difetti alla nascita e aborto in gravidanza clinicamente riconosciuta è rispettivamente dal 2 al 4% e dal 15 al 20%.
Dati
Dati sugli animali
Le scimmie in gravidanza di Cynomolgus sono state somministrate cetuximab per via endovenosa una volta alla settimana durante il periodo di organogenesi (Giornata della gestazione [GD] 20-48) a livelli di dose da 0,4 a 4 volte la dose raccomandata di Erbitux basata sulla superficie corporea (BSA). Il cetuximab è stato rilevato nel fluido amniotico e nel siero di embrioni da dighe trattate su GD 49. Mentre non si sono verificate malformazioni fetali nella prole, si è verificata una maggiore incidenza di embrioletalità e aborti a dosi di circa 1 a 4 volte la dose raccomandata di ERbitux basata su BSA.
Nei topi l'EGFR è di fondamentale importanza nei processi riproduttivi e di sviluppo tra cui lo sviluppo della placenta dell'impianto di blastocisti e la sopravvivenza e lo sviluppo embrione-feteo/postnatale. La riduzione o l'eliminazione della segnalazione EGFR embrione o materna può prevenire l'impianto può causare la perdita di embrioni-fetali durante le varie fasi della gestazione (attraverso gli effetti sullo sviluppo della placenta) e può causare anomalie dello sviluppo e morte precoce nei feti sopravvissuti.
Risultati avversi dello sviluppo sono stati osservati in più organi negli embrioni/neonati di topi con segnalazione EGFR interrotta.
Lattazione
Riepilogo del rischio
Non ci sono informazioni sulla presenza di Erbitux nel latte umano Gli effetti sul bambino allattato al seno o sugli effetti sulla produzione di latte. Gli anticorpi IgG umani possono essere escreti nel latte umano. A causa del potenziale di gravi reazioni avverse nei bambini allattati al seno di Erbitux consiglia alle donne di non allattare durante il trattamento con Erbitux e per 2 mesi dopo l'ultima dose di Erbitux.
Femmine e maschi di potenziale riproduttivo
Gravidanza Testing
Verificare lo stato di gravidanza nelle femmine del potenziale riproduttivo prima di iniziare l'erbitux [vedi Usa in una popolazione specifica ].
Contraccezione
Sulla base del suo meccanismo d'azione, Erbitux può causare danni al feto quando somministrato a una donna incinta [vedi Utilizzare in popolazioni specifiche ].
Femmine
Consiglio alle femmine del potenziale riproduttivo di utilizzare una contraccezione efficace durante il trattamento con ERBITUX e per 2 mesi dopo l'ultima dose di Erbitux.
Infertilità
Femmine
Sulla base degli studi sugli animali, ERBEX può compromettere la fertilità nelle femmine di potenziale riproduttivo [vedi Tossicologia non clinica ].
Uso pediatrico
La sicurezza e l'efficacia dell'erbitux nei pazienti pediatrici non sono stati stabiliti. La farmacocinetica di cetuximab in combinazione con irinotecan è stata valutata in pazienti pediatrici con tumori solidi refrattari in uno studio di dose di dose a braccio singolo openlabel. Erbitux è stato somministrato una volta una settimana a dosi fino a 250 mg/m² a 27 pazienti che vanno da 1 a 12 anni; e in 19 pazienti che vanno dai 13 ai 18 anni. Non sono stati identificati nuovi segnali di sicurezza nei pazienti pediatrici. La farmacocinetica del cetuximab tra le due fasce di età era simile a seguito di una singola dose di 75 mg/m² e 150 mg/m². Il volume della distribuzione sembra essere indipendente dalla dose e approssima lo spazio vascolare da 2 L/m² a 3 L/m². A seguito di una singola dose di 250 mg/m², l'AUC0-INF media (CV%) era di 17,7 mg*H/mL (34%) nella fascia di età più giovane (1 € 12 anni n = 9) e 13,4 mg*H/ml (38%) nel gruppo adolescenziale (13 € 18 anni n = 6). L'emivita media di cetuximab era di 110 ore (da 69 a 188 ore) nel gruppo più giovane e 82 ore (da 55 a 117 ore) nel gruppo adolescenziale.
Uso geriatrico
Dei 1662 pazienti con carcinoma del colon-retto avanzato che hanno ricevuto erbitux con irinotecan con folfiri o come singleagente in sei studi (Bond IMCL-CP02-9923 IMCL-CP02-0141 IMCL-CP02-0144 CA225-025 e cristallo) 35% dei pazienti erano 65 anni di età o più vecchi. Non sono state osservate differenze complessive nella sicurezza o nell'efficacia tra questi pazienti e i pazienti più giovani.
Studi clinici sull'erbitux condotto in pazienti con carcinoma della testa e del collo non includevano un numero sufficiente di soggetti di età pari o superiore a 65 anni per determinare se rispondono in modo diverso dai soggetti più giovani.
Dei 216 pazienti con mutazione BRAF V600E CRC metastatico positivo che ha ricevuto Erbitux in combinazione con Encorafenib 300 mg una volta al giorno il 29% aveva 65 anni fino a 75 anni mentre 20 (9%) avevano 75 anni e oltre [vedi oltre [vedi oltre [vedi oltre [vedi oltre [vedi oltre [vedi oltre [vedi Studi clinici ].
Non sono state osservate differenze complessive nella sicurezza o nell'efficacia di Erbitux più encorafenib nei pazienti anziani rispetto ai pazienti più giovani.
Informazioni per overdose per Erbitux
Nessuna informazione fornita
Controindicazioni per Erbitux
Nessuno.
Farmacologia clinica for Erbitux
Meccanismo d'azione
Il recettore del fattore di crescita epidermico (EGFR HER1 C-ERBB-1) è una glicoproteina transmembrana che è membro di una sottofamiglia della tirosina chinasi del recettore di tipo I, incluso EGFR HER2 HER3 e HER4. L'EGFR è costitutivamente espresso in molti tessuti epiteliali normali tra cui la pelle e il follicolo pilifero. L'espressione di EGFR è rilevata anche in molti tumori umani, compresi quelli del colon della testa e del collo e del retto.
è verapamil un bloccante del canale di calcio
Il cetuximab si lega specificamente all'EGFR sia sulle cellule normali che tumorali e inibisce in modo competitivo il legame del fattore di crescita epidermico (EGF) e altri ligandi come la trasformazione del fattore di crescita alfa. I test in vitro e gli studi sugli animali in vivo hanno dimostrato che il legame del cetuximab ai blocchi EGFR blocca la fosforilazione e l'attivazione delle chinasi recettorassociate con conseguente inibizione dell'induzione della crescita cellulare dell'apoptosi e una riduzione della metalloproteinasi della matrice e della produzione di matrice vascolare. La trasduzione del segnale attraverso l'EGFR provoca l'attivazione di proteine RAS di tipo selvaggio, ma in cellule con attivazione di mutazioni somatiche di RAS Le risultanti proteine Mutanti RAS sono continuamente attive indipendentemente dalla regolazione dell'EGFR.
Il cetuximab in vitro può mediare la citotossicità cellulare anticorpale-dipendente (ADCC) contro alcuni tipi di tumore umano. Saggi in vitro e studi sugli animali in vivo hanno dimostrato che il cetuximab inibisce la crescita e la sopravvivenza delle cellule tumorali che esprimono l'EGFR. Non sono stati osservati effetti antitumorali di cetuximab negli xenotrapianti tumorali umani privi di espressione di EGFR. L'aggiunta di cetuximab alla radioterapia o irinotecan nei modelli di xenotrapianto tumorale umano nei topi ha comportato un aumento degli effetti anti-tumore rispetto alla sola radioterapia o chemioterapia.
Nell'ambito dell'induzione CRC mutante BRAF dell'attivazione della via MAPK mediata da EGFR è stata identificata come un meccanismo di resistenza agli inibitori BRAF. È stato dimostrato che le combinazioni di un inibitore BRAF e agenti mirano all'EGFR superano questo meccanismo di resistenza nei modelli non clinici. La somministrazione di co-cetuximab e encorafenib hanno avuto un effetto antitumorale maggiore di entrambi i farmaci da solo in un modello murino di carcinoma del colon-retto con BRAF V600E mutata.
Farmacocinetica
L'erbitux somministrato come singolo agente o in combinazione con chemioterapia concomitante o radioterapia presenta farmacocinetici non lineari. L'area sotto la curva del tempo di concentrazione (AUC) è aumentata in modo proporzionale maggiore che dose, mentre la clearance di cetuximab è diminuita da 0,08 l/h/m² a 0,02 l/h/m² mentre la dose è aumentata da 20 mg/m² a 200 mg/m² e plateauEd a dosi> 200 mg/m².
L'esposizione sistemica di cetuximab dopo la somministrazione di Erbitux era del 22% (IC 90%: 6% 38%) superiore a quella di un altro prodotto Cetuximab utilizzato in estremo e cristallo.
Distribuzione
Il volume della distribuzione per cetuximab sembrava essere indipendente dalla dose e approssimava lo spazio vascolare di 2 € 3 l/m².
Eliminazione
In seguito al dosaggio raccomandato (400 mg/m² dose iniziale; 250 mg/m² di dose settimanale) di cetuximab ha raggiunto i livelli di stato stazionario dalla terza infusione settimanale con concentrazioni di picco e depressione attraverso gli studi che vanno da 168 μg/ml a 235 μg/ml e 41 μg/ml a 85 μg rispettivamente. L'emivita media di cetuximab era di circa 112 ore (da 63 a 230 ore).
Popolazione specifica
La funzione epatica e renale della razza sessuale di età non ha avuto effetti clinicamente significativi sulla farmacocinetica del cetuximab. La clearance di cetuximab è aumentata di 1,8 volte man mano che l'area della superficie corporea è aumentata da 1,3 m² a 2,3 m², il che è coerente con il dosaggio raccomandato di cetuximab su base mg/m².
Studi sull'interazione farmacologica
Non è stata osservata alcuna interazione farmacocinetica tra cetuximab e irinotecan cetuximab e cisplatino e cetuximab e carboplatino.
Non sono state osservate differenze clinicamente significative nella farmacocinetica di cetuximab o encorafenib quando la dose iniziale di Erbitux raccomandata di 400 mg/m² è stata somministrata con Encorafenib.
Studi clinici
Carcinoma a cellule squamose della testa e del collo (SCCHN)
In combinazione con la radioterapia
Bonner (NCT00004227) era uno studio randomizzato controllato multicentrico di 424 pazienti con SCCHN a livello locale o regionale. I pazienti con SCCHN in stadio III/IV dell'orofaringe ipofaringe o laringe senza terapia precedente sono stati randomizzati (1: 1) per ricevere l'erbitux in combinazione con la sola radioterapia o radioterapia. I fattori di stratificazione sono stati lo stato di prestazione di Karnofsky (60 € 80 contro 90 € 100) stadio nodale (N0 contro N) stadio tumorale (T1 € 3 contro T4 utilizzando i criteri di stadiazione dell'American Joint on Cancer 1998) e la frazione di terapia con radiazione (rammentlazione concomitante rispetto a una volta-Daily contro due volte faily). La radioterapia è stata somministrata per 6 settimane di 7 settimane come una spinta Oncedily due volte al giorno o concomitante. L'erbitux è stato somministrato per via endovenosa come una dose iniziale di 400 mg/m² a partire da una settimana prima dell'inizio della radioterapia seguita da 250 mg/m² a settimana somministrata 1 ora prima della radioterapia per la durata della radioterapia (6 € 7 settimane). La principale misura di risultato di efficacia era la durata del controllo locoregionale. Un'altra misura di risultato è stata la sopravvivenza globale (OS).
Dei 424 pazienti randomizzati, l'età media era di 57 anni l'80% era maschio L'83% era bianco e il 90% aveva lo stato di prestazione di Karnofsky al basale ≥80. C'erano 258 pazienti arruolati nei siti statunitensi (61%). Il sessanta per cento dei pazienti harofaringeo al 25% laringeo e tumori primari ipofaringei al 15%; Il 28% aveva stadio tumorale AJCC T4. Il cinquantasei per cento dei pazienti ha ricevuto radioterapia con un aumento concomitante del 26% ha ricevuto regime una volta al giorno e il 18% del regime due volte al giorno.
I risultati di efficacia sono presentati nella Tabella 8.
Tabella 8: Efficacia Risultati in SCCHN locoregionalmente avanzato in Bonner
| Erbitux più radiazioni (n = 211) | Solo radiazioni (n = 213) | Hazard Ratio (95% CIa) | Valore p di log-rank stratificato | |
| Controllo locoregionale | ||||
| Durata mediana (mesi) | 24.4 | 14.9 | 0.68 (0,52-0,89) | 0.005 |
| Sopravvivenza globale | ||||
| Durata mediana (mesi) | 49.0 | 29.3 | 0.74 (0,57-0,97) | 0.03 |
| a CI = intervallo di confidenza. |
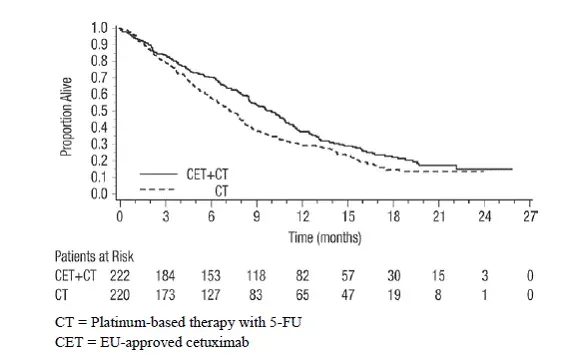
In combinazione con terapia a base di platino con fluorouracile
Extreme (NCT00122460) era uno studio multicentrico randomizzato in aperto di 442 pazienti con malattia locoregionale ricorrente o SCCHN metastatico. I pazienti con terapia precedente per la malattia locoregionale ricorrente o lo SCCHN metastatico sono stati randomizzati (1: 1) per ricevere un prodotto cetuximab in combinazione con terapia a base di platino e fluorouracile o terapia a base di platino e solo fluorocile. La scelta del cisplatino o del carboplatino era a discrezione dell'investigatore. I fattori di stratificazione erano Karnofsky Stato delle prestazioni (<80 versus ≥80) and previous chemioterapia. Cisplatin (100 mg/m² intravenously on Day 1) or carboplatin (AUC 5 mg/mL*min intravenously on Day 1) and fluorourEil (1000 mg/m²/day intravenously on Days 1â€4) were administered every 3 weeks (1 cycle) for a maximum of 6 cycles in the absence of disease progression or unEceptable toxicity. Cetuximab was administered intravenously at a 400 mg/m² initial dose followed by a 250 mg/m² weekly dose. In the absence of disease progression or unEceptable toxicity Diter completion of 6 planned courses of platinum-based therapy weekly cetuximab as a single-agent could be continued until disease progression or unEceptable toxicity. If chemioterapia was delayed because of adverse reEtions weekly cetuximab was continued. If chemioterapia was discontinued for adverse reEtions weekly cetuximab as a single-agent could be continued until disease progression or unEceptable toxicity. The main efficEy outcome measure was OS. Altro outcome measures were PFS and objective response rate (Orr).
Dei 442 pazienti randomizzati, l'età media era di 57 anni il 90% era maschio il 98% era bianco e l'88% aveva lo stato di prestazione di Karnofsky al basale ≥80. Il trentaquattro per cento dei pazienti aveva una cavità orale orofaringea del 25% laringea e tumori primari ipofaringei. Il cinquantatre per cento dei pazienti aveva solo una malattia locoregionale ricorrente e il 47% aveva una malattia metastatica. Il cinquantotto per cento aveva una malattia in stadio IV AJCC e il 21% aveva una malattia in stadio III. Il sessantaquattro per cento dei pazienti ha ricevuto terapia con cisplatino e il 34% ha ricevuto carboplatino come terapia iniziale. Circa il quindici percento dei pazienti nel solo braccio di cisplatino è passato al carboplatino durante il periodo di trattamento.
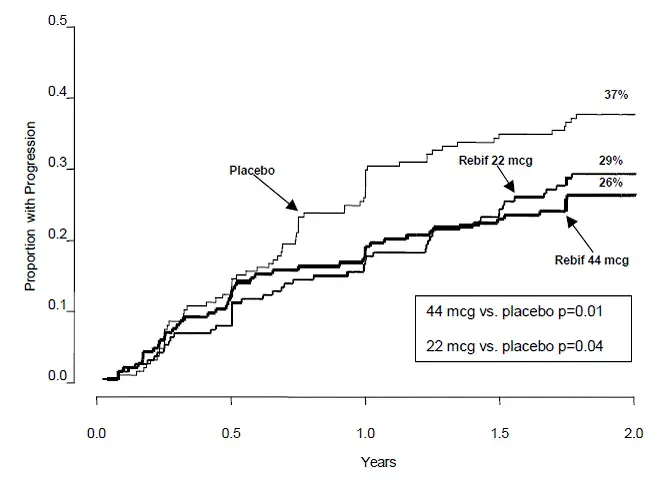
I risultati di efficacia sono presentati nella Tabella 9 e nella Figura 1.
Tabella 9: l'efficacia si traduce in una malattia locoregionale ricorrente o SCCHN metastatico in Extreme
| Cetuximab con terapia a base di platino e fluorouracile (n = 222) | Terapia a base di platino e fluorocile (n = 220) | |
| Sopravvivenza globale | ||
| Durata mediana (mesi) | 10.1 | 7.4 |
| Hazard Ratio (95% CIa) | 0,80 (0,64 0,98) | |
| Valore p di log-rank stratificato | 0.034 | |
| Sopravvivenza libera da progressione | ||
| Durata mediana (mesi) | 5.5 | 3.3 |
| Hazard Ratio (95% CIa) | 0,57 (0,46 0,72) | |
| Valore p di log-rank stratificato | <0.0001 | |
| Tasso di risposta obiettivo | 35,6% | 19,5% |
| Odds ratio (IC al 95%a) | 2.33 (1.50 3.60) | |
| CMHb Test p-valore | 0.0001 | |
| a CI = intervallo di confidenza. b CMH = Cochran-Mantel-HManszel. |
Figura 1: curve Kaplan-Meier per la sopravvivenza globale in pazienti con malattia locoregionale ricorrente o SCCHN metastatico in Extreme
 |
Nelle analisi esplorative del sottogruppo mediante terapia con platino iniziale (cisplatino o carboplatino) per i pazienti (n = 284) che ricevevano cetuximab in combinazione con cisplatino e fluorocile rispetto al cisplatino e al fluorocile da solo. La differenza nella PFS mediana era di 2,1 mesi (5,6 contro 3,5 mesi; HR 0,55; IC 95% 0,41 0,73). L'ORR era rispettivamente del 39% e del 23% (OR 2,18; IC 95% 1,29 3,69).
Per i pazienti (n = 149) che ricevono cetuximab in combinazione con carboplatina e fluorouracile rispetto al carboplatino e al fluorouracile da sola la differenza nel sistema operativo mediano era di 1,4 mesi (9,7 contro 8,3 mesi; HR 0,99; 95% IC 0,69 1,43). La differenza nella PFS mediana era di 1,7 mesi (rispettivamente 4,8 contro 3,1 mesi; HR 0,61; IC 95% 0,42 0,89). L'ORR era rispettivamente del 30% e del 15% (OR 2,45; IC 95% 1,10 5,46).
Come singolo agente
EMR 62202-016 era uno studio clinico multicentrico a braccio singolo in 103 pazienti con SCCHN ricorrente o metastatico. Tutti i pazienti avevano documentato la progressione della malattia entro 30 giorni da un regime di chemioterapia a base di platino. I pazienti sono stati somministrati per via endovenosa una dose di test di 20 mg di Erbitux il giorno 1 seguito da una dose iniziale di 400 mg/m² e 250 mg/m² settimana fino a una progressione della malattia o una tossicità inaccettabile.
L'era mediana era di 57 anni l'82% era bianco al 100% e il 62% aveva uno stato di performance di Karnofsky di ≥80.
L'ORR era del 13% (IC 95% 7% 21%). La durata mediana della risposta (DOR) è stata di 5,8 mesi (intervallo da 1,2 a 5,8 mesi).
K-RAS wild wild-type che esprime il carcinoma del colon-retto metastatico (CRC)
In combinazione con folfiri
Il cristallo (NCT00154102) era uno studio multicentrico randomizzato di etichette aperte su 1217 pazienti con MCRC che esprime l'EGFR. I pazienti sono stati randomizzati (1: 1) a ricevere un prodotto cetuximab in combinazione con Folfiri o Folfiri da solo come trattamento di prima linea. I fattori di stratificazione erano lo stato di prestazione del gruppo di oncologia cooperativa orientale (ECOG) (0 e 1 contro 2) e regione (Europa occidentale contro l'Europa orientale rispetto ad altri).
FOLFIRI regimen included 14-day cycles of irinotecan (180 mg/m² intravenously on Day 1) folinic acid (400 mg/m² [racemic] or 200 mg/m² [L-form] intravenously on Day 1) and fluorouracil (400 mg/m² bolus on Day 1 followed by 2400 mg/m² as a 46-hour continuous infusion). Il cetuximab è stato somministrato per via endovenosa come una dose iniziale di 400 mg/m² seguita da 250 mg/m² a settimana somministrata 1 ora prima della chemioterapia. Il trattamento dello studio è continuato fino alla progressione della malattia o alla tossicità inaccettabile. La principale misura di risultato di efficacia è stata valutata da PFS da un comitato di revisione indipendente (IRC). Altre misure di esito erano OS e ORR.
Dei 1217 pazienti randomizzati, l'età media era di 61 anni il 60% era maschio L'86% era bianco e il 96% aveva uno stato di performance ECOG basale 0 € 1 60% aveva un tumore primario localizzato nel colon l'84% aveva 1 € 2 siti metastatici e il 20% aveva ricevuto precedenti adiuvanti e/o neoadjuvant chemiterapia. I dati demografici e le caratteristiche di base erano simili tra le armi di studio.
Lo stato di mutazione K-Ras era disponibile per l'89% dei pazienti: il 63% aveva tumori di tipo selvaggio K-RAS e il 37% aveva tumori mutanti K-Ras in cui i test valutati per le seguenti mutazioni somatiche nei codoni 12 e 13 (esone 2): G12A G12D G12R G12R G12C G12S G12V G13D. Le caratteristiche di base e i dati demografici nel sottoinsieme di tipo selvaggio K-RAS erano simili a quelle osservate nella popolazione complessiva.
È stato osservato un miglioramento statisticamente significativo della PFS per il cetuximab con braccio Folfiri rispetto al braccio Folfiri (PFS mediano 8,9 contro 8,1 mesi HR 0,85 [IC 95% 0,74 0,99] Value p = 0,036). L'OS non era significativamente diverso all'analisi finale pianificata basata su 838 eventi (HR = 0,93 IC 95% [0,8 1,1] Value p 0,327).
Risultati dell'analisi PFS e ORR pianificata in tutti i pazienti randomizzati e analisi PFS e ORR post-hoc nei sottogruppi di pazienti definiti dallo stato di mutazione K-RAS e dall'analisi post-hoc di OS aggiornato in base a ulteriori follow-up (1000 eventi) in tutti i pazienti randomizzati e nei sottogruppi dei pazienti definiti da K-RAS sono stati presentati da un effetto per il trattamento per il trattamento per il trattamento. pazienti con tumori di tipo selvaggio K-RAS. Non ci sono prove di efficacia nel sottogruppo di pazienti con tumori mutanti K-Ras.
Tabella 10: l'efficacia si traduce nel carcinoma del colon-retto metastatico che esprime l'EGFR di prima linea in cristallo (tutto lo stato randomizzato e K-RAS)
| Tutto randomizzato | K-Ras wild-type | Mutante K-Ras | ||||
| Cetuximab con folfiri (n = 608) | Folfiri (n = 609) | Cetuximab con folfiri (n = 320) | Folfiri (n = 356) | Cetuximab con folfiri (n = 216) | Folfiri (n = 187) | |
| Sopravvivenza libera da progressione | ||||||
| Numero di eventi (%) | 343 (56) | 371 (61) | 165 (52) | 214 (60) | 138 (64) | 112 (60) |
| Mediano (mesi) (95% CI) | 8.9 (8.0 9.4) | 8.1 (7.6 8.8) | 9.5 (8.9 11.1) | 8.1 (7.4 9.2) | 7.5 (6.7 8.7) | 8.2 (7.4 9.2) |
| Hr (95% CI) | 0.85 (0,74 0,99) | 0.70 (0,57 0,86) | 1.13 (0,88 1,46) | |||
| valore pa | 0.0358 | |||||
| Sopravvivenza globaleb | ||||||
| Numero di eventi (%) | 491 (81) | 509 (84) | 244 (76) | 292 (82) | 189 (88) | 159 (85) |
| Mediano (mesi) (95% CI) | 19.6 (18 21) | 18.5 (17 20) | 23.5 (21 26) | 19.5 (17 21) | 16.0 (15 18) | 16.7 (15 19) |
| Hr (95% CI) | 0.88 (0,78 1.0) | 0.80 (0,67 0,94) | 1.04 (0,84 1,29) | |||
| Tasso di risposta obiettivo | ||||||
| Orr (95% CI) | 46% (42 50) | 38% (34 42) | 57% (51 62) | 39% (34 44) | 31% (25 38) | 35% (28 43) |
| a In base al test log-rank stratificato. b Risultati dell'analisi del sistema operativo post-hoc basati su altri 162 eventi. |
Figura 2: curve di Kaplan-Meier per la sopravvivenza globale nella popolazione di tipo selvaggio K-Ras in cristallo
 |
Come singolo agente
Lo studio CA225-025 (NCT00079066) è stato uno studio clinico randomizzato con etichette open multicentrici condotta in 572 pazienti con MCRC ricorrente precedentemente trattato con EGFR. I pazienti sono stati randomizzati (1: 1) a ricevere ERBITUX con la migliore cure di supporto (BSC) o la sola BSC. L'erbitux è stato somministrato per via endovenosa come una dose iniziale di 400 mg/m² seguita da 250 mg/m² a settimana fino alla progressione della malattia o alla tossicità inaccettabile. La principale misura di risultato di efficacia era il sistema operativo. Dei 572 pazienti randomizzati, l'età media era di 63 anni il 64% era maschio L'89% era bianco e il 77% aveva uno stato di prestazione ECOG basale di 0 € 1. I dati demografici e le caratteristiche di base erano simili tra le armi di studio. Tutti i pazienti dovevano aver ricevuto e progredito in terapia precedente tra cui un regime contenente IRinotecan e un regime contenente ossaliplatino.
Lo stato K-Ras era disponibile per il 79% dei pazienti: il 54% aveva tumori di tipo selvaggio K-RAS e il 46% aveva tumori mutanti K-Ras in cui i test valutati per le seguenti mutazioni somatiche nei codoni 12 e 13 (Exone 2): G12A G12D G12R G12R G12C G12S G12V G13D.
I risultati di efficacia sono presentati nella Tabella 11 e nella Figura 3.
Tabella 11: sopravvivenza globale nel carcinoma del colon-retto metastatico che esprime EGFR precedentemente trattato nello studio CA225-025 (tutto lo stato randomizzato e K-RAS)
| Tutto randomizzato | K-Ras wild-type | Mutante K-Ras | ||||
| Erbitux con BSC (N = 287) | BSC (N = 285) | Erbitux con BSC (N = 117) | BSC (N = 128) | Erbitux con BSC (N = 108) | BSC (N = 100) | |
| Mediano (mesi) (95% CI) | 6.1 (5.4 6.7) | 4.6 (4.2 4.9) | 8.6 (7.0 10.3) | 5.0 (4.3 5.7) | 4.8 (3.9 5.6) | 4.6 (3.6 4.9) |
| Hr | 0.77 | 0.63 | 0.91 | |||
| (95% CI) | (0,64 0,92) | (0,47 0,84) | (0,67 1,24) | |||
| valore pa | 0.0046 | |||||
| a In base al test log-rank stratificato. |
Figura 3: Curve Kaplan-Meier per la sopravvivenza globale in pazienti con carcinoma del colon-retto metastatico di tipo selvaggio K-RAS nello studio CA225-025
 |
In combinazione con irinotecan
Il legame era uno studio clinico multicentrico condotto in 329 pazienti con MCRC ricorrente che espresse EGFR. I campioni tumorali non erano disponibili per il test per lo stato di mutazione K-Ras. I pazienti sono stati randomizzati (2: 1) a ricevere ERBETUX in combinazione con Irinotecan (218 pazienti) o singolo agente Erbitux (111 pazienti). L'erbitux è stato somministrato per via endovenosa come una dose iniziale di 400 mg/m² seguita da 250 mg/m² a settimana fino alla progressione della malattia o alla tossicità inaccettabile. Nell'erbitux con il braccio di Irinotecan, l'Iinotecan è stato aggiunto all'erbitux usando lo stesso dosaggio per irinotecan che il paziente aveva precedentemente fallito. I programmi di irinotecan accettabili erano 350 mg/m² ogni 3 settimane 180 mg/m² ogni 2 settimane o 125 mg/m² settimanali di quattro dosi ogni 6 settimane. L'efficacia di Erbitux con singolo agente Irinotecan o Erbitux basato su risposte oggettive durevoli è stata valutata in tutti i pazienti randomizzati e in due sottopopolazioni pre-specificate: fallimenti refrattari e irinotecan e ossaliplatino irinotecan.
Dei 329 pazienti l'età media era di 59 anni il 63% era maschio il 98% era bianco e l'88% aveva lo stato di prestazione di Karnofsky al basale ≥80. Circa i due terzi avevano precedentemente fallito il trattamento di ossaliplatino.
Nei pazienti che ricevevano erbitux con irinotecan l'ORR era del 23% (IC 95% 18% 29%) DOR mediana era di 5,7 mesi e il tempo mediano alla progressione era di 4,1 mesi. Nei pazienti che ricevevano Erbitux come singolo agente, l'ORR era dell'11% (IC 95% 6% 18%) DOR mediana era di 4,2 mesi e il tempo mediano alla progressione era di 1,5 mesi. Sono stati osservati tassi di risposta simili nei sottoinsiemi predefiniti sia nel braccio di combinazione che nel braccio a agente singolo.
BRAF V600E Mutation-Positive Metastatic Cancer (CRC)
L'erbitux in combinazione con Encorafenib è stato valutato in uno studio multicentrico randomizzato controllato a etichetta aperta (Beacon CRC; NCT02928224). I pazienti idonei dovevano avere CRC metastatico positivo a mutazione BRAF V600E, rilevato utilizzando il kit di reazione a catena della polimerasi RGQ di Qiagen Therascreen BRAF V600E con progressione della malattia dopo 1 o 2 regimi precedenti. Altri criteri di ammissibilità chiave includevano l'assenza di un precedente trattamento con un'idoneità dell'inibitore RAF MEK o EGFR di ricevere cetuximab per etichettatura locale rispetto allo stato RAS tumorale e allo stato delle prestazioni ECOG (PS) 0â € 1. La randomizzazione è stata stratificata dallo stato delle prestazioni ECOG (0 contro 1) l'uso precedente di Irinotecan (sì contro NO) e il prodotto CeTuximab utilizzato (licenziato USA contro approvato dall'UE).
I pazienti sono stati randomizzati 1: 1: 1 a uno dei seguenti bracci di trattamento:
- Erbitux in combinazione con encorafenib 300 mg per via orale una volta al giorno (braccio Erbitux/Encorafenib)
- Erbitux in combinazione con binimetinib e encorafenib 300 mg per via orale una volta al giorno
- Erbitux con irinotecan o erbitux con folfiri (control arm)
Il dosaggio di cetuximab in tutti i pazienti era di 400 mg/m² per via endovenosa per la prima dose seguita da 250 mg/m² a settimana.
I pazienti nel braccio di controllo hanno ricevuto erbitux con Irinotecan 180 mg/m² per via endovenosa nei giorni 1 e 15 di ogni ciclo di 28 giorni o folfiri per via endovenosa (Irinotecan 180 mg/m² nei giorni e 15; acido folinico 400 mg/m con giorni 1 e 15; allora fluouracil 400 MG/m² su giorni di fluoura di Fluoura mg /m² /giorno per infusione continua per 2 giorni).
Il trattamento è continuato fino alla progressione della malattia o alla tossicità inaccettabile. Di seguito sono descritti solo i risultati del regime approvato (Erbitux in combinazione con Encorafenib).
La principale misura di risultato di efficacia era OS. Ulteriori misure di esito dell'efficacia includevano PFS ORR e DOR valutati da Blinded Independent Central Review (BICR). OS e PFS sono stati valutati in tutti i pazienti randomizzati. ORR e DOR sono stati valutati nel sottoinsieme dei primi 220 pazienti inclusi nella parte randomizzata dell'erbitux/encorafenib e del braccio di controllo dello studio.
Un totale di 220 pazienti sono stati randomizzati al braccio Erbitux/Encorafenib e 221 al braccio di controllo. Di questi 441 pazienti l'età media era di 61 anni; Il 53% era femmina; L'80% era bianco e il 15% era asiatico. Il cinquanta percento (50%) aveva uno stato di performance ECOG basale di 0; Il 66% ha ricevuto 1 terapia precedente e il 34% ha ricevuto 2; Il 93% ha ricevuto oxaliplatino precedente e il 52% ha ricevuto precedente IRINOTECAN.
Erbitux in combinazione con Encorafenib ha dimostrato un miglioramento statisticamente significativo in OS ORR e PFS rispetto al comparatore attivo. I risultati di efficacia sono riassunti nella Tabella 12 e nella Figura 4.
Tabella 12: L'efficacia risulta nel carcinoma del colon-retto BRAF V600E mutazione positivo in Beacon CRC
| Erbitux con encorafenib N = 220 | Erbitux con irinotecan o erbitux con folfiri N = 221 | |
| Sopravvivenza globale | ||
| Numero di eventi (%) | 93 (42) | 114 (52) |
| Mediano OS months (95% CI) | 8.4 (7,5 11.0) | 5.4 (4.8 6.6) |
| Hr (95% CI)ab | 0,60 (0,45 0,79) | |
| valore pE | 0.0003 | |
| Tasso di risposta complessivo (per BICR) | ||
| Orr (95% CI)d | 20% (13% 29%) | 2% (0% 7%) |
| Cr | 5% | 0% |
| Pr | 15% | 2% |
| valore pMa | <0.0001 | |
| Mediano DoR months (95% CI) | 6.1 (4.1 8.3) | No (2.6 no) |
| Sopravvivenza libera da progressione ( per BICR) | ||
| Numero di eventi (%) | 133 (60) | 128 (58) |
| Malattia progressiva | 110 (50) | 101 (46) |
| Morte | 23 (10) | 27 (12) |
| Mediano PFS months (95% CI) | 4.2 (3.7 5.4) | 1.5 (1.4 1.7) |
| Hr (95% CI)ab | 0,40 (0,31 0,52) | |
| valore pDi | <0.0001 | |
| Ci = intervallo di confidenza; Cr = risposta completa; Dor = durata della risposta; HR = rapporto hazard; Nr = non raggiunto; ORR = tasso di risposta complessivo; OS = sopravvivenza globale; PFS = sopravvivenza libera da progressione; PR = risposta parziale. a Stratificato dalla fonte ECOG PS di cetuximab (autorizzazione USA contro approvata dall'UE) e precedente l'uso di Irinotecan alla randomizzazione. b Modello di pericolo proporzionale Cox stratificato. c Test di log-rank stratificato testato a livello alfa di 0,0084. d ARM ERBITUX/ENCORAFENIB (n = 113) e braccio di controllo (n = 107). e Test di Cochran-Mantel-Haenszel; Testato a livello di alfa di 0,05. f Test di log-rank stratificato testato a livello alfa di 0,0234. |
Figura 4: Curve di Kaplan-Meier per la sopravvivenza globale in pazienti con carcinoma del colon-retto BRAF V600E mutazione positivo
Informazioni sul paziente per Erbitux
Reazione di infusiones
Consiglia ai pazienti che il rischio di gravi reazioni di infusione può essere aumentato nei pazienti che hanno avuto un morso di zecche o un'allergia alla carne rossa. Consiglia ai pazienti di contattare il proprio operatore sanitario e di segnalare segni e sintomi delle reazioni di infusione, comprese le reazioni di infusione a esordio tardivo come i brividi della febbre o i problemi di respirazione [vedi AVVERTIMENTOS AND PRECAUZIONI ].
Arresto cardiopolmonare
Consiglia ai pazienti il rischio di arresto cardiopolmonare o morte improvvisa e per segnalare qualsiasi storia di malattia coronarica insufficienza cardiaca congestizia o aritmie [vedi AVVERTIMENTOS AND PRECAUZIONI ].
Polmonare Toxicity
Consiglia ai pazienti di contattare immediatamente il proprio operatore sanitario per il dolore al torace per la tosse nuovo o peggioramento o la mancanza di respiro [vedi AVVERTIMENTOS AND PRECAUZIONI ].
Dermatologico Toxicities
Consiglia ai pazienti di limitare l'esposizione al sole durante il trattamento Erbitux e per 2 mesi dopo l'ultima dose di Erbitux. Consiglia ai pazienti di avvisare il proprio operatore sanitario di qualsiasi segno di eruzione cutanea simile all'acne (che può includere prurito secco secco o cracking pelle e infezione da infiammazione o gonfiore alla base delle unghie o perdita delle unghie) congiuntivite blefarite o una visione ridotta [vedi visione AVVERTIMENTOS AND PRECAUZIONI ].
Tossicità dell'embrione-fetale
Consiglia a pazienti di sesso femminile di potenziale riproduttivo del potenziale rischio per un feto e per utilizzare una contraccezione efficace durante il trattamento ERbitux e per 2 mesi dopo l'ultima dose di Erbitux. Consiglia ai pazienti di informare il proprio operatore sanitario di una gravidanza nota o sospetta [vedi AVVERTIMENTOS AND PRECAUZIONI Utilizzare in popolazioni specifiche ].
Lattazione
Consiglia ai pazienti di non allattare al seno durante il trattamento Erbitux e per 2 mesi dopo l'ultima dose di Erbitux [vedi Utilizzare in popolazioni specifiche ].